
丙二酸溶液中的铝阳极氧化研究
丙二酸溶液中的铝阳极氧化研究
任建军 左 禹
(北京化工大学材料科学与工程学院,北京100029)
摘要:在丙二酸电解液中进行了铝阳极氧化,并采用扫描电镜(SEM)、X射线衍射(XRD)及显微硬度仪对氧化膜的构型、物质结构、耐酸性、显微硬度等进行了表征。结果表明,丙二酸电解液的黏度随浓度的升高而增大,电解液黏度较大时,氧化膜的生长速率会明显降低;电流密度较大、氧化时间较长时能显著提高氧化膜构型的规整度;氧化膜膜厚的增长速率呈现出先增大后降低的规律;在相同的氧化条件下,电解液浓度较大时,得到的氧化膜的厚度也较大。在800℃以上对丙二酸氧化膜加热,原来无定型的氧化铝会结晶生成![]() -Al2O3;氧化膜结晶后其耐酸性会显著提高。丙二酸电解液的浓度对氧化膜的显微硬度没有显著影响,氧化膜硬度对膜厚的依赖存在一个极限值,约为27µm。
-Al2O3;氧化膜结晶后其耐酸性会显著提高。丙二酸电解液的浓度对氧化膜的显微硬度没有显著影响,氧化膜硬度对膜厚的依赖存在一个极限值,约为27µm。
引 言
除草酸外,对于在其他有机羧酸溶液中的阳极氧化还相当少见,这可能与有机羧酸的自身性质有关。与无机酸相比,有机羧酸的酸性相对较低,并且多数羧酸难溶于水。所以尽管有机羧酸种类较多,但适合阳极氧化工艺要求的却很少。继草酸之后,丙二酸是酸性最强的二元羧酸,并且其溶解度比草酸要大得多,因此丙二酸是为数不多的适合阳极氧化工艺要求的有机羧酸之一。当然一些羟基羧酸如酒石酸、柠檬酸等也能用来制备多孔型的阳极氧化膜。
关于丙二酸阳极氧化的研究,至今已有一些成果报道。丙二酸阳极氧化膜的生长电压较高,可达120 V左右,其单元尺寸较大,达300 nm左右。阳极氧化过程中羧酸根阴离子会在氧化膜中形成夹杂,这些夹杂的阴离子会形成特色的发光中心,故丙二酸阳极氧化膜与无机酸阳极氧化膜相比具有特殊的光学性质。将丙二酸阳极氧化膜加热到500~600℃时,夹杂的羧酸根阴离子会发生脱羧反应而产生CO2。
总之,对于丙二酸阳极氧化,虽然很早就受到个别研究者的关注,但被公布的研究成果仍然较少。因此,本文以丙二酸阳极氧化体系为研究对象,从氧化膜生长的电化学行为、自组织行为以及氧化膜的性能等各个方面进行系统的探索,以便进一步深化对丙二酸阳极氧化的认识。
1 实验部分
1.1 实验原料
1.2铝阳极氧化膜的制备
在丙酮中对高纯铝试片超声波清洗15 min,除去表面的油脂,然后用去离子水充分冲洗,并用冷风吹干。对脱脂的铝试片在0. 48、1.92及3.84mol/L3种不同浓度的丙二酸水溶液中进行阳极氧化。用MPS 705直流稳压电源(北京切克斯公司)作为阳极氧化的供电设备,采取恒电压、恒电流两种氧化模式,在双电极电解池中进行,以面积比铝试片大得多的石墨板做为电解池的阴极。氧化过程中将电解液温度恒定在14℃,并对电解液施加强烈搅拌。在恒电压氧化模式下,将电压设定在100 V,氧化过程一直持续到铝试片基本被完全氧化、电流显示为“0”为止。在恒电流氧化模式下,电流密度及氧化时间依实验条件而定。氧化结束后将试样从电解池中取出,用离子水充分冲洗,并用冷风吹干。
1.3 测试与表征
用日本日立场发射扫描电子显微镜(S4700型)对氧化膜的形貌进行观察,并对氧化膜的成分进行EDS分析。用日本理学D/max2500型X射线衍射仪(XRD)对丙二酸氧化膜的晶型进行表征。为研究丙二酸氧化膜的高温结晶行为,将氧化膜试样在马福炉中高温加热(830℃)4 h,并进行XRD表征。在磷酸溶液中(将黏稠的磷酸加入到等体积的水中)对丙二酸氧化膜进行浸泡(50℃),研究氧化膜的耐酸性,浸泡时间依实验条件确定。将浸泡过的氧化膜试样用去离子充分冲洗,并用冷风吹干。用场发射扫描电子显微镜对浸泡过的氧化膜试样进行形貌观察。
2 结果与讨论
2.1 丙二酸阳极氧化的电化学行为
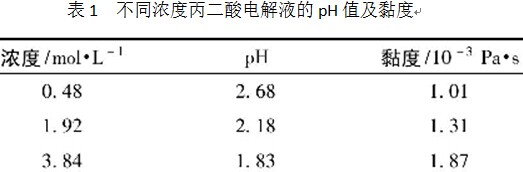
图1为3种不同浓度的丙二酸电解液中恒电流氧化模式下电压一时间变化曲线,氧化电流密度为10 mA/cm2,电解液浓度分别为0. 48、1.92及3.84mol/L。可见3条曲线特征相似,描述的均是多孔型氧化膜的生长过程。对上述3种不同浓度的丙二酸电解液进行pH值和黏度测定,结果如表1所示。
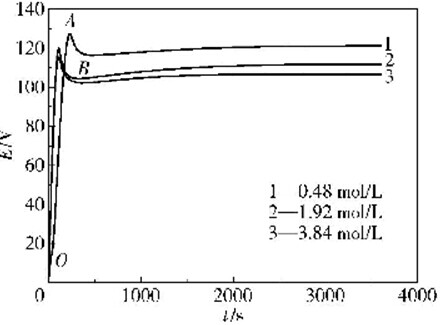
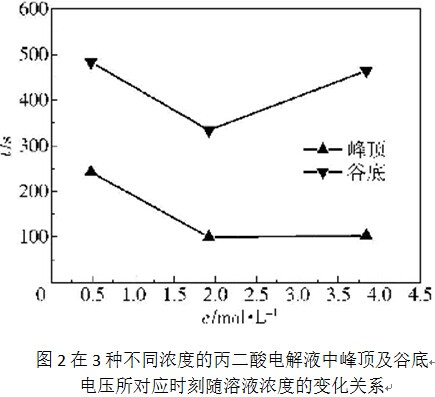
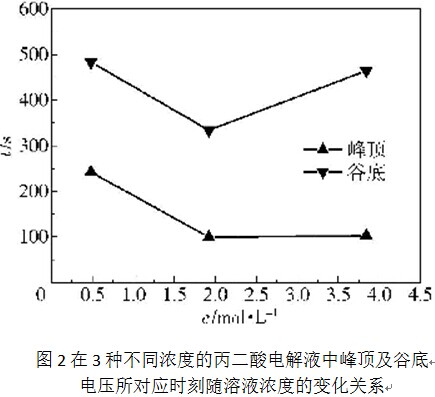
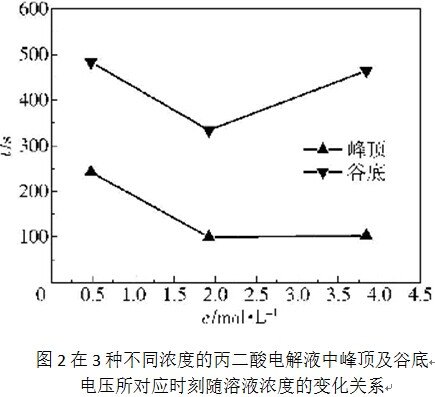
在这3种不同浓度的丙二酸电解液中,氧化膜的生长电压上升到峰顶(如图1中的A点)及下降至谷底(如图1中的B点)所对应的时刻与电解液浓度的变化关系如图2所示。可见,随着电解液浓度从0. 48mol/L增加到1.92mol/L,电压上升至峰顶及下降至谷底所对应的时刻均在显著降低;但随着电解液浓度从1. 92mol/L进一步增大到3.84mol/L,这两个时刻又有所增大,尤其是谷底电压时刻增大的更为显著。与此相对应,第一暂态阶段(如图1所示的OA段)直线段的斜率随着电解液浓度的增大呈现出先显著增大后略微降低的情形。以上结果表明,随着电解液浓度从1. 92mol/L进一步增大到3. 84mol/L,氧化膜的生长速率有所减小,这可能是因为电解液黏度进一步增大的结果。电解液黏度增大会使离子的扩散传质变得困难,进而会降低阻挡层中的离子迁移速率及氧化膜的生长速率。
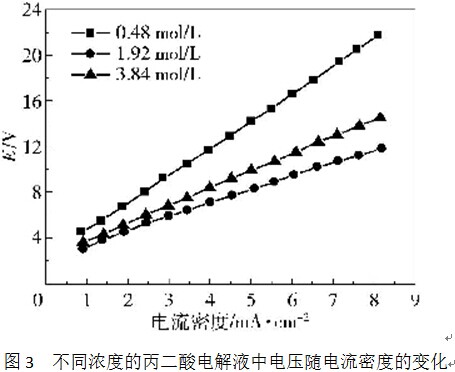
在玻璃电解池中对丙二酸电解液的导电性进行了测定。以面积相等的两块石墨板作为阳极与阴极,通以较小电流,以防止发生剧烈的电极反应。在实验过程中电极表面没有发生明显的气泡析出。然后记录各个电流密度下的稳定电压,对电压-电流密度进行做图,研究二者之间的变化关系,结果如图3所示。
从图3可看出,在3种不同浓度的丙二酸电解液中电压随电流密度的变化均符合线性关系,并且直线的斜率ξ呈现如下的变化规律:ξ0.48>ξ3.84>ξ1.92,也即电解液浓度从0.48mol/L增加到1.92mol/L,其导电性随之增加;从1.92mol/L进一步增加到3. 84mol/L,其导电性又有所降低,这可能也是基于电解液黏度显著增大的结果。
从以上实验结果可得知,对于类似丙二酸等有机羧酸电解液,因其酸性较低,通常需要采用较大的浓度;但浓度增大同时又会引起溶液黏度的显著增大,会产生不利于氧化膜生长的反向效应,因此需要合理控制电解液的浓度。
2.2 丙二酸阳极氧化膜的构型
2.2.1恒电压氧化模式
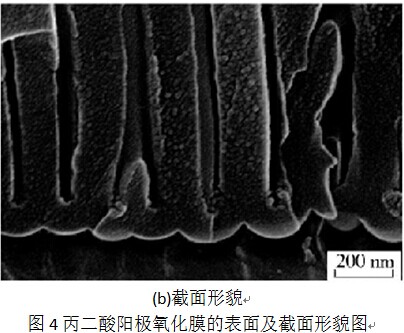
对20 µm厚的高纯铝试样在0.48mol/L的丙二酸电解液中进行恒电压阳极氧化,氧化电压设定在100 V,进行双面氧化,氧化时间约为10 h。生成的氧化膜呈现出完全透明的状态,颜色为金黄色,金属基体己基本被完全消耗。氧化膜厚度约为30 µm,表明与消耗的金属基体相比变厚了,厚度比约为1.5。在氧化过程中电流密度始终维系在大约4mA/cm2的较小数值。氧化膜的生长速率较小,约为0. 42 nm/s。氧化膜的形貌如图4所示。从图4(a)可看出,膜孔清晰地暴露在表面,但相邻的孔间距不很均匀,平均值为200 nm左右,并且膜孔的形状也不太规则,表明氧化膜的构型较差。还可以看到,氧化膜表面除暴露的膜孔外,还分布着许多浅坑,表明氧化膜表面发生了明显的化学溶解。可见,0. 48mol/L的丙二酸电解液尽管酸性很小,但在长达10h的氧化过程中,也能够对氧化膜产生明显的化学溶解,但氧化膜表面及膜孔并未遭到严重的破坏,仍旧维持着非常完好的状态。氧化膜的截面形貌如图4(b)所示,可见氧化膜也是由一个个柱状单元密排构成,单元尺寸比较大,但构型不规整。
对生成的丙二酸阳极氧化膜进行EDS分析,发现氧化膜中除含有Al、O两种元素外,还含有明显的C。C、O、Al各原子分数为3.96%、56. 52%及39. 52%,表明在氧化过程中溶液中的羧酸根阴离子- OOC—CH2—COO -在氧化膜中形成了明显的夹杂。假定氧化膜由Al2O3和Al2(CH2(COO)2)3组成,根据原子分数,可以估算出氧化膜的平均组成为Al2O3 ·0.022 Al2(CH2(COO)2)3。

2.2.2恒电流氧化模式
如图4所示,在电流密度较小及膜层较薄的情况下,氧化膜的构型较差。据文献报道,氧化电流密度(或电场强度)是影响氧化膜构型或自组织行为的关键因素,电流密度越大,构型越规整,此外延长氧化时间也能显著改善氧化膜的构型。因此,为深入探讨氧化膜的自组织行为,以便能够显著提高氧化膜的构型,就在比较优化的条件下进行丙二酸阳极氧化膜的制备。图2的实验结果表明,在1. 92mol/L的丙二酸电解液中氧化膜的生长速率较大,并且溶液的黏度也不大,因此就将电解液的浓度选定在1. 92mol/L;采取恒电流氧化模式,将电流密度提高至21mA/cm2;将氧化时间延长至8h以上,采取单面氧化的方式。在氧化过程中,氧化膜的稳定生长电压达到了120 V左右,生成的氧化膜的厚度约为370µm。氧化膜的形貌如图5所示。从图5(a)可看出,氧化膜构型表现出了较高的规整度,大部分单元呈现出规则的六边形形状,相互之间密集排列在一起,单元尺度比较均匀,单元的平均中心距(或孔间距)约为313 nm。与图4相比,可见电流密度增大、氧化膜生长电压提高后,孔间距发生了显著的增大。氧化膜的截面形貌如图5(b)所示。可看出膜孔的孔道很直,每个单元独立发展,没有出现连孔现象,并且孔间距分布非常均匀,但也存在孔径大小不一的缺陷。但总的来看,氧化膜的自组织程度比较高,其构型的规整度较为可观。


2.3 丙二酸阳极氧化膜的生长速率
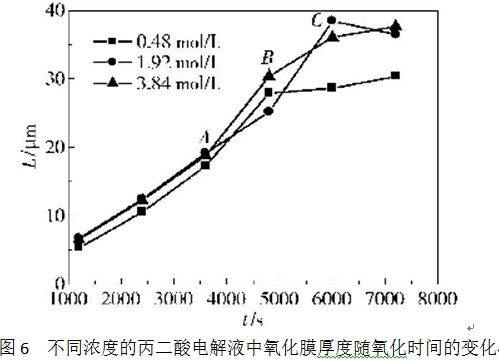
图6描述了在3种不同浓度的丙二酸电解液中氧化膜厚度L随时间的变化关系,氧化电流密度为12mA/cm2。用扫描电镜对氧化膜的厚度进行测定。从图中可以看出,在3种不同浓度的丙二酸电解液中,氧化膜的生长速率均表现出先逐渐增大然后逐渐降低的规律。以浓度为3. 84mol/L丙二酸电解液所对应的曲线为例进行分析,可以看出,在氧化膜的整个生长过程中,AB段所体现的生长速率最大,假定在BC段继续保持同样的生长速率,则在C点氧化膜的厚度应为42 µm,而实际的膜厚只有36 µm,也即氧化膜的膜厚损失了6 µm。但这不应归因于丙二酸电解液的化学溶解作用,因为丙二酸溶液的酸性较弱,在较短的时间内不可能对膜厚产生微米数量级的溶解。因此,氧化膜生长速率的降低另有其他原因。随着氧化膜的生长,膜孔孔道在逐渐延长,孔道会限制其中溶液的自由流动,从而使得离子的扩散传质受到阻碍,孔道越长这种阻碍作用越大,进而会降低阻挡层中的离子电流,结果使得氧化膜生长速率相应地减小。
从3条曲线总的特征来看,在3. 84mol/L的丙二酸电解液中,氧化膜的生长过程最为稳定;在1. 92mol/L的丙二酸电解液中,氧化膜的生长过程略有波动,但与3. 84mol/L丙二酸电解液中的生长情况还是比较接近的;在0. 48mol/L的丙二酸电解液中,后期氧化膜的生长有点受阻。该实验结果表明,尽管3. 84mol/L的丙二酸电解液黏度较大,对氧化膜的生长有负面影响,但其较大的酸性还是有利于氧化膜稳定生长的。同时还可以看出,在这3种不同浓度的丙二酸电解液中,在各个时刻氧化膜的厚度是有差别的。相比之下,在0. 48mol/L的丙二酸电解液中氧化膜最薄。氧化膜所体现的这种膜厚差异可能与酸根阴离子在膜中的夹杂有关。由于酸根阴离子的夹杂,使得氧化膜与纯净的Al2O3相比,体积有所膨胀;夹杂越严重,膨胀越显著。
2.4 丙二酸阳极氧化膜的晶态结构及性能
2.4.1 晶态结构
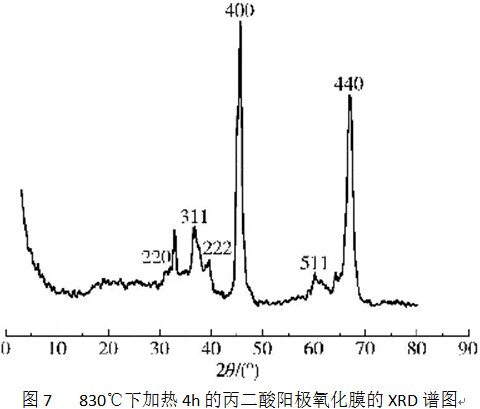
XRD分析表明,外观呈金黄色的丙二酸阳极氧化膜为无定型的氧化铝。但是在适当的高温条件下,丙二酸氧化膜会发生结晶。图7为830℃下加热4h的丙二酸阳极氧化膜的XRD谱图。图中出现了γ-Al2O3的特征峰,表明原来无定型的氧化铝结晶生成了γ-Al2 O3至少是部分结晶)。对于硫酸阳极氧化膜来说,当温度达到970℃时,原来无定型的氧化铝也会结晶为γ-Al2O3;温度继续升高到1100℃时,γ -Al2O3又转变为δ-Al2O3;当达到1200℃时,全部转变为稳定的α-Al2O3。对于草酸阳极氧化膜来说,当温度达到820℃时,无定型的Al2O3开始结晶为γ-Al2O3,到840℃时,几乎全部转变为γ-Al2O3;随着温度的持续升高又相继转变为δ-Al2O3和θ-Al2O3;直到1150℃完全转变为稳定的α-Al2O3。可见,在高温条件下发生结晶或相转变是铝阳极氧化膜的一个共同特征。
实验中发现,当丙二酸阳极氧化膜经高温加热结晶后,其耐酸性会显著提高,结果如图8所示。图8(a)为原始制备的氧化膜在磷酸溶液中浸泡3 min后的表面形貌。图8(b)为经高温热处理发生结晶的氧化膜在磷酸溶液中浸泡70 min后的表面形貌。图8(a),(b)的氧化膜均是在1. 92mol/L的丙二酸电解液中10 mA/cm2的电流密度下经120 min氧化所得。如图8(a)所示,氧化膜表面被溶解的痕迹非常明显,膜孔显著扩大,孔径超过了100 nm,孔壁遭受到了剧烈的溶解。相比之下,图8(b)所示的氧化膜非常完好,孔径没有明显扩大,基本维持原来的形貌,看不出发生明显的溶解。可见经高温加热结晶后,丙二酸氧化膜的耐酸性显著提高。
2.4.3 显微硬度
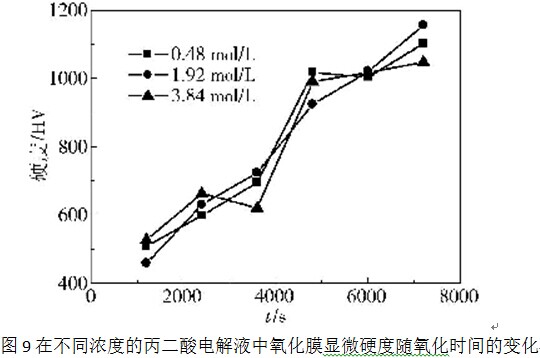
图9为3种不同浓度的丙二酸电解液中,氧化膜维氏硬度随氧化时间的变化关系,氧化电流密度为10 mA/cm2。从图中可以看出,电解液浓度对氧化膜的显微硬度没有明显的影响,氧化膜硬度随膜厚的增大存在一个极限值,这个极限厚度约对应80 min的氧化时间,膜厚应为27 µm左右。这可能是因为氧化膜达到这一厚度时,多孔层的孔型及膜的构型就达到了相对稳定的程度,因此对外力的承载能力也就基本确定。


 3结论
3结论
(1)丙二酸电解液的黏度随浓度的升高而增大,电解液黏度较大时,会显著降低氧化膜的生长速率;丙二酸电解液对氧化膜的化学溶解作用较弱;电流密度较大、氧化时间较长时能显著提高氧化膜构型的规整度。
(2)丙二酸阳极氧化,膜厚的增长速率呈现出先增大后降低的规律;其他氧化条件相同时,在较浓的丙二酸电解液中生成的氧化膜厚度也较大。
(3)在800℃以上对丙二酸氧化膜加热时,原来无定型的氧化物会结晶生成γ-Al2 O3;氧化膜结晶后耐酸性会显著提高。
(4)电解液浓度对氧化膜显微硬度没有明显影响;氧化膜硬度对膜厚的依赖存在一个极限厚度值。
