
浅议铝阳极氧化理论
浅议铝阳极氧化理论
吴双成
(甘肃皋兰胜利机械有限公司热处理,甘肃 皋兰730200)
[摘要] 简述了铝阳极氧化工艺的发明历史和理论发展,对传统阳极氧化理论进行了分析推理,提出不同看法,尤其是阳极界面溶液的pH值问题,认为pH值应该在7.8~10.8范围之间,“碱致溶解”是形成多孔层的原因。
阳极氧化是指铝及其合金的表面处理方法,可以制得高品质的防护、装饰和功能性膜。通过阳极氧化方法制备的阳极氧化铝薄膜分为壁垒型和多孔型两种。通常情况下,壁垒型是在中性电解液中形成的,而多孔型是在酸性(如磷酸、草酸等)电解液中形成的,传统理论认为壁垒型和多孔型的生长机制不同。
1 阳极氧化工艺和理论的发展简史
早在1896年,波拉克Pollak就提出了在硼酸或磷酸溶液中直流电解,可得到“堡垒”型氧化膜的专利。1923年英国的本戈Bengough和斯图尔特Stuart发明铬酸阳极氧化,1923年,日本的Kujirai和植木Ueki首先发明了草酸阳极氧化法,在日本应用较为广泛,其后德国也相继出现了这种处理工艺,电解液基本成分是质量分数2%~10%的草酸溶液,使用直流电或交流电。1927年,高尔Gower、奥布莱思·斯塔福德Stafford OBrien和Partners4发表了硫酸阳极氧化的专利,氧化的电流密度为0.7~1. 3A/dm2,这种电流密度一直延用到现在。1937年,在英国首先用硫酸阳极氧化法对铝的表面进行电化学处理,对铝合金制品装饰、保护和表面硬化。1982年保加利亚人几B.Tpq×npoB提出,选用三乙醇胺做为稳定添加剂,与盐类联合使用,能得到青铜色、黄色至咖啡色的彩色铝阳极氧化膜。1990年美国波音公司的G.M. Wong等人取得硼酸-硫酸阳极氧化专利,该工艺对电源要求低,氧化速度快,节省能源,能够取代铬酸阳极氧化,成为环保型氧化工艺。
自1923年阳极氧化工艺问世以来,许多研究者都对其形成机理和组成结构进行了研究。1953年Keller等人提出了铝阳极氧化膜的六角柱模型,又称物理-几何模型或Keller模型。这个模型精辟的解释了铝阳极氧化膜的性质,曾经被公认是铝阳极氧化膜生成机理的权威性理论。这个理论把膜的溶解仅仅看作是一种纯粹的化学溶解,1961年,Murphy等人为了解决“六角柱模型”在解释电流恢复现象时,不能解释不同电压条件下溶解阻挡层速度相差很大的问题,提出了“质子空间电荷”的概念。认为阳极上铝的氧化应由下述任何一种反应引起:
2Al+3H2O → Al2O3 +6H+ +6e
2A1+3OH → Al2O3 +3H+ +6e
2Al+3HSO4- → Al2(SO4)3 +3H+ +6e
这些反应都能释放出游离态质子。上世纪60年代末,0.Sullivan、G.C.Wood等人提出,阻挡层的溶解除了除了化学溶解之外,还有电化学溶解。永山等人进一步指出,即便在电流停滞期间,孔底的溶解仍然主要是电场溶解,孔壁的溶解主要依靠化学溶解,电场溶解速度要远大于化学溶解速。Cherki等人在硫酸电解液中用氧的同位素18O和16O作为示踪原子,对氧的反应历程进行了研究,发现在电场作用下O2和Al3+在Al2O3膜层中扩散速度都比较快,而且O2-的扩散速度快于Al3+。O2-扩散到氧化膜内部与Al3+结合而成Al2O3,O2-是在氧化膜与溶液界面上生成的。
2 “酸致溶解理论”简介
“酸致溶解理论”得到了表面处理界多数人的公认,有几十年的历史,可以说是“传统”阳极氧化理论。“酸致溶解理论”简述如下:
将铝件在硫酸、铬酸、磷酸、草酸等电解质溶液中,作阳极进行电解处理,则在其表面形成较厚的氧化膜,膜的主要成分是Al2O3。过去认为,在酸性溶液中,水的电解生成初生态氧,初生态氧具有很强的氧化能力,它与阳极铝作用生成氧化膜,并且放出大量的热,即
H2O-2e → [O]+2H+
2Al+3[O] → Al2O3 +1669J
Al2O3 +H2O → Al2O3·H2O
这就是成膜作用,在氧化膜形成过程中,同时还要发生膜的化学溶解,也就是同时发生溶膜作用,即
2Al+6H+ → 2Al3+ +3H2↑
Al2O3 +6H+ → 2Al3+ +3H2O
对硫酸阳极氧化来说,溶膜物质是硫酸,对草酸阳极氧化来说,溶膜物质是草酸。阳极氧化膜的生长是在膜的生成和溶解这一矛盾过程中发生和发展的。
阳极氧化过程的实质是H+与OH+的放电和接着进行新生态氧对铝的氧化,形成Al2O3。氧化开始时,阳极上很快地生成一层薄而致密的氧化膜(即紧贴金属表面的无孔内层),电阻也大,在最初的几秒钟里就使电压急剧升高,电压升高导致氧化膜局部薄的地方“击穿”。击穿的地方膜的溶解加快,出现孔穴,并继而延伸,为膜的进一步增厚提供条件,而且以孔为中心形成一个个六角柱形组成的氧化膜的外层。
由氧化铝所形成的内层氧化膜组织密实,硬度高,厚度约为0.01~0.1µm,具有很高的绝缘电阻,这层氧化膜不易被电解液渗透,所以称为阻挡层,由玻璃状的无水Al2O3组成的,是γ- Al2O3;与电解液接触的外层氧化膜是多孔层,主要组成是无水Al2O3和Al2O3·H2O,常温下为非晶态Al2O3,高温下为Al2O3晶体,其次组成是酸或酸分解产物等,如在硫酸电解液中所得膜含有少量Al2(SO4)3及SO3,这层氧化膜具有不少孔隙,易被电解液渗透,在通电时,就不断地使膜层的微孔增多、增深和变厚,最终达到厚度1~200µm。
3 对“酸致溶解理论”的异议
阳极氧化机理比较复杂,氧化膜是通过什么样的历程生成的?反应究竟有几步?至今还有许多问题没有弄清楚。有关多孔型氧化膜的形成机理已经研究了50多年,传统理论认为“酸性场致溶解”是氧化膜表面孔洞产生和孔道发展的主要原因,但是“酸性场致溶解”理论对多孔性的六棱柱元胞和半球形底部结构无法做出合理解释。铝在进行阳极氧化时,存在电解液/氧化物和氧化物/Al两个界面,传统理论认为壁垒型是在这两个界面同时生长的,而多孔型是在氧化物/Al界面上生长,而在电解液/氧化物界面发生“酸性场致溶解”导致孔洞的形成和加深。对此朱绪飞认为多孔型规则孔道是“氧气气泡模具效应”的结果。
(1)传统的“酸致溶解理论”是没有经过实验验证的理论,查遍文献资料也没有具体测定阳极界面pH值的研究报道。既然阳极界面附近溶液是酸性的,应该有详细研究报告,内容包括谁测定的?用什么方法、什么仪器测定的?测定的结果是多少?等等。所以,“酸致溶解理论”是没有经过科学证实的理论,还是一种处于假设阶段的理论。
(2)按照“酸致溶解理论”,阳极在电解H2O的同时会释放出H+,使阳极界面附近溶液的酸性增强,应该比溶液本体的酸性更强。假如硫酸阳极氧化溶液中,硫酸的质量浓度是196g/L,即2mol/L,那么,铝阳极界面的酸性一定大于2mol/L,这么强的酸性,氧化膜Al2O3会被全部溶解,如同酸洗一样,铝表面不会存在氧化膜。
(3)“酸致溶解理论”没有考虑阳离子、阴离子的迁移速度、放电速度问题。H+和OH-离子半径小,虽然迁移速度快,但是它们的放电还原氧化速度却不是很快,在铝阳极界面往往会出现OH-的积累而碱化,阳极电流密度越大,积累的OH -浓度就会越高。
(4)传统“酸致溶解理论”中,对阳极氧化时的热效应数据不够准确,本人曾经做过探讨,应该为2Al+3[O] → Al2O3+1669KJ。铝氧化是一个放热反应,氧化膜生成时在铝基体表面会产生很多的热量,其生成热为15. 91KJ/m2。
(5)传统“酸致溶解理论”中,阳极溶解膜的反应2Al+6H+ → 2Al3+ +3H2↑是不能成立的,阳极只有氧气析出,不会有氢气出现。
(6)不可能单独存在O2-,一定是以某种形式存在,笔者认为是以OH-形式存在的。
既然是一种理论,就应该能够解释该工艺的大部分问题,然而“酸致溶解理论”对以上问题是无法解释的。因此,本人对“传统”阳极氧化理论——“酸致溶解理论”早有怀疑,提出了“碱致溶解理论” 假设。
4 “碱致溶解理论假设”简介
铝及铝合金的阳极氧化同电抛光有相近的阳极过程。氧化膜的生成和溶解是同时进行的,在阳极表面可能发生以下几种反应:
(1)铝离子溶入到电解液中Al - 3e= Al3+
(2)氧化膜的生成Al+3OH- - 3e → Al(OH)3
(3)在电热量作用下
2Al(OH)3 → (Al2O3·H2O +2H2O) → Al2O3 +3H2O
(4)气态氧的析出4OH- - 4e =2H2O +O2↑
(5)电解液中其它组分在阳极表面上的氧化;
(6)小孔处氧化膜的溶解
Al(OH)3+OH- → AlO2- +2H2O
(7)氧化膜的再生成
2AlO2- +2H+ → Al2O3·H2O
阳极氧化膜的生成和溶解交替进行,周而复始。
(1)离子的电迁移:阳极表面带有正电荷,吸引溶液中的OH-离子和酸根阴离子,OH-离子被阳极表面氧化为氧气或原子氧;阴极表面带有负电荷,吸引溶液中的H+,H+被阴极表面还原为氢气或原子氢。
OH-离子和酸根阴离子通过小孔向铝基体阳极方向迁移,而阳极产生的氧气从铝基体通过小孔向外逸出,H+跑向阴极并还原为氢气逸出。
(2)阳极界面的酸碱性:OH-离子半径小,电迁移速度很陕,H+和OH-离子在25℃无限稀溶液中的电迁移速度分别是3. 63×10-7和2.052 xl0-7m2·S-1·V-1,但是OH-离子的放电氧化速度却较慢,在铝阳极界面OH-离子的补充速率大于消耗速率,会出现OH-的积累而碱化,阳极电流密度越大,积累的OH-浓度就会越高。
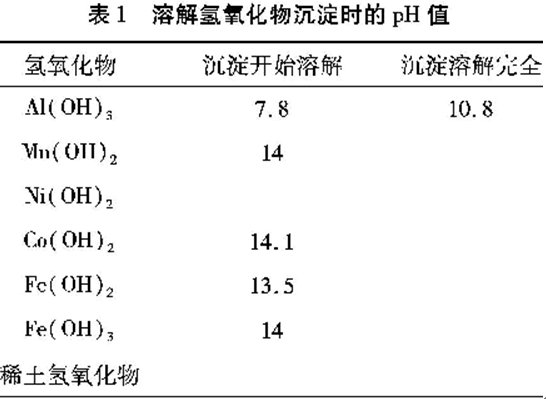
Al(OH)3沉淀开始溶解的pH值为7.8,沉淀完全溶解的pH值为10.8。如果阳极界面pH值低于7.8,则Al(OH)3沉淀就不会被OH-离子溶解,氧化膜过于致密阻止氧化膜的继续生成,也不符合阳极氧化膜存在多孔层的事实。从氧化开始时,阳极上很快地生成一层薄而致密的氧化膜(即紧贴金属表面的无孔内层),电阻也大,在最初的几秒钟里就使电压急剧升高,电压升高导致氧化膜局部薄的地方“击穿”,随后电压迅速降低的实际现象看,Al(OH)3沉淀开始溶解了,因此,阳极界面pH值大于7.8。
如果阳极界面pH值大于10.8,则Al(OH)3沉淀就会全部被OH-离子溶解,没有氧化膜存在。铝阳极氧化膜由致密的内层(阻挡层)和多孔的外层(疏松层)组成。
因此,阳极界面溶液不是呈强酸性,而是呈弱碱性,pH值应该在7.8~10.8范围之间。在氧化膜内层pH≈7.8,外层pH≈10.8,从外层到内层pH值递减。
(3)阳极界面的电流密度:从表面上看阳极氧化时阳极的平均电流密度为1~2A/dm2,由于氧化膜具有绝缘性,使得电流只是在阳极氧化膜的孔隙处导电。阳极氧化膜小孔所占面积约为总面积的10%~15%,开孔率是10%~15%,故小孔处的电流密度为平均电流密度与开孔率的比值。
当平均电流密度等于1A/dm2时,小孔处的电流密度为1÷10%=10A/dm2或1÷15%=6.67A/dm2;当平均电流密度等于2A/dm2时,小孔处的电流密度为2÷10%=20A/dm2或2÷15%=13.33A/dm2。
总地来说,小孔处电流密度大约在6.67A/dm2~20A/dm2之间,电流密度是相当高的。如此高的电流密度使得小孔处的阳极极化相当强,足以氧化大部分物质。
5 “碱致溶解理论假设”的应用
2005年我国表面处理界老专家郑瑞庭先生,在《电镀与精饰》刊物第一期上提出,铝件阳极氧化时出现粉红色液体是什么物质?由何引起?本人尝试利用“碱致溶解理论假设”解释了这一问题,解释基本上成功。但是当时没有考虑到阳极氧化溶液中,可能添加硫酸锰、硝酸锰等“导电盐”的实际情况,以及溶液使用过久,可能积累了较多的Mn2+因素。总之,可以肯定溶液中含有Mn2+,才会出现这种现象。
假若阳极氧化溶液中既存在Mn2+,又存在草酸,是不是阳极表面会看不到粉红色呢?答案是也能看到红色现象。因为,阳极界面将Mn2+氧化成MnO4后,阳极界面呈弱碱性,碱性条件下MnO4-氧化能力弱,无法氧化草酸,也就是说,碱性条件下草酸不能还原MnO4-,使MnO4-离子的粉红色消失。只有MnO4-离开阳极表面进入溶液本体,在酸性条件下才能被还原成无色Mn2+。
符德学等人在研究锌锰合金的电沉积时,同样发现溶液中的Mn2+不稳定,当电流密度增大到一定值时,Mn2+易被阳极氧化为MnO2甚至MnO4-。阳极区镀液发红,且颜色随电流密度的增大而加深,用分光光度法测得525nm处有MnO4-的特征吸收峰,证明有MnO4-生成。说明了在阳极放出氧气的同时,二价锰离子也会被氧化。这就是得到实际证明的例子。
解释铝阳极氧化时的“导电盐”:铝阳极氧化中,加入MnSO4、Mn(NO3)2、NiSO4、CoSO4、草酸钴、FeSO4、稀土化合物如磺基水杨酸镧铈等,能够提高溶液的导电能力,为“导电盐”的观点是令人怀疑的,这些盐的导电能力还不如硫酸。适量的这些物质,能够降低铝氧化膜的孔隙率,提高铝氧化膜的硬度,加快铝氧化膜的生成速度。这与它们的耐碱性有关,这些金属离子与阳极界面的OH-反应,生成氢氧化物后,与Al(OH)3相比不容易被过量的OH-再溶解,致使氧化膜空隙率下降,氧化膜显得致密、硬度提高、生成速度加快。
解释铝阳极氧化时的其它添加剂的作用:由于氧化膜的溶解反应Al(OH)3+OH- → AlO2- +2H2O是个吸热反应,温度高时有利于溶膜反应,所以,温度越高对氧化膜的溶解越快,OH-浓度越高越容易溶解氧化膜。这两种因素都会使氧化膜孔隙增多增大,降低氧化膜硬度和耐蚀性,降低成膜速度。混酸硬质常温氧化中加入的有机酸,如草酸、丙二酸、乳酸、苹果酸、磺基水杨酸、酒石酸等,在阳极表面上被氧化时需要吸收热量,阻止阳极界面温度的上升,减弱OH-对膜的溶解,使氧化膜孔隙少、硬度和抗腐蚀性提高。甘油热容量大,不容易上升温度,也能起到一定的减弱氧化膜的溶解反应的作用。硼酸是典型的路易斯酸,能够加合一个OH-,缓和阳极界面上OH-浓度的上升,防止阳极界面附近溶液pH值上升较快,减弱OH-对氧化膜的溶解造成硬度低、孔隙率高、成膜速度慢。
人们对阳极的极化过程研究不多,阳极的电化学反应了解不够,今后有条件者应该加强这方面的研究。
